The increased consumption of fossil resources is responsible for the emission of large amounts of anthropogenic CO2, which results in climate change and ocean acidification. Direct conversion of carbon dioxide (CO2) using green hydrogen is a sustainable approach to jet fuel production. However, achieving a high level of performance remains a challenge due to the inertness of CO2 and its low activity for subsequent C–C bond formation.
Motivated by such a formidable challenge, a research team led by Prof. Gao Peng, Prof. Li Shenggang and Prof. Sun Yuhan at Shanghai Advanced Research Institute (SARI) of the Chinese Academy of Sciences reported a Na-modified CoFe alloy catalyst using layered double-hydroxide precursors that directly transforms CO2 to a jet fuel composed of C8-C16 jet-fuel-range hydrocarbons with very high selectivity. The research results were published in The Innovation entitled "Direct conversion of CO2 to a jet fuel over CoFe alloy catalysts”.
The Na-modified CoFe alloy catalyst demonstrates a highly efficient production of jet-fuel-range hydrocarbons with an unprecedentedly high C8-C16 selectivity of 63.5% with 10.2% CO2 conversion as well as a low combined selectivity of less than 22% toward undesired CO and CH4 via direct CO2 hydrogenation (Figure 1).
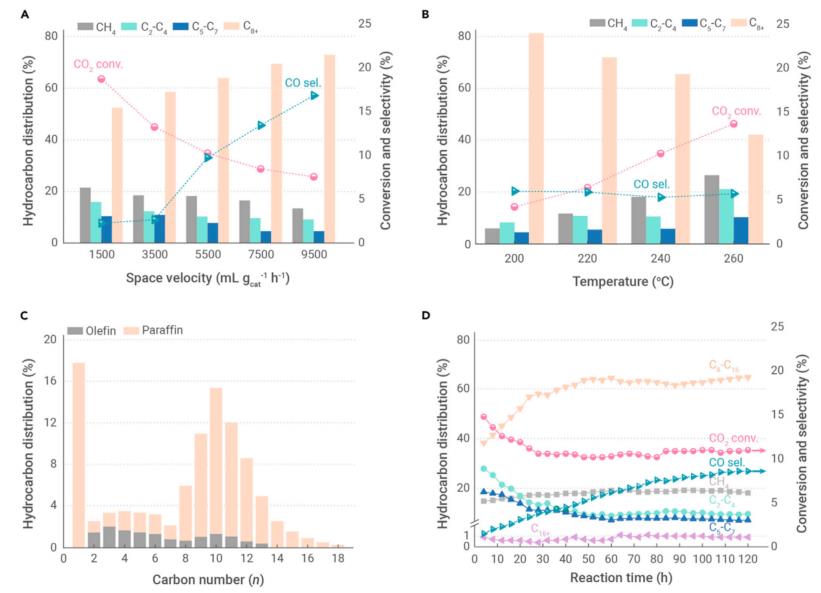
Figure 1. The evaluation data of CO2 hydrogenation over CoFe-0.81Na catalyst. Effects of (A) space velocity and (B) reaction temperature on conversion of CO2, selectivity of CO, and hydrocarbon distribution (Image by SARI)
Structural characterization reveals that the Co7Fe3 alloy structure inhibits CH4 formation and plays a crucial role in the selectivity hydrogenation of CO2 to the higher hydrocarbons that constitute jet fuel.
The spectroscopic studies suggest that the CoFe alloy surface has a remarkable inhibitory effect on CH4 production via CO2 methantion and the metallic CoFe alloy is the active phase responsible for producing C2+ hydrocarbons from the CO intermediate, whose formation is facilitated by the iron oxide surface sites generated in situ during the CO2 hydrogenation reaction.
Density functional theory calculations show that the CoFe catalyst surface has a lower tendency for the C-C coupling reactions than the Co catalyst surface, as reflected from both the calculated energy barriers and reaction energies (Figure 2). This contributes to the narrower carbon distribution from hydrogenation of the CO intermediate over the CoFe catalyst, compared to the Co catalyst.
The calculations further show that formation of the CoFe alloy suppresses the CO2 methanation reaction by increasing the energy barrier for the dissociation of the CH3O* intermediate, leading to a lower methane selectivity than that over the Co catalyst. These theoretical predictions are in good agreement with our experimental observations.
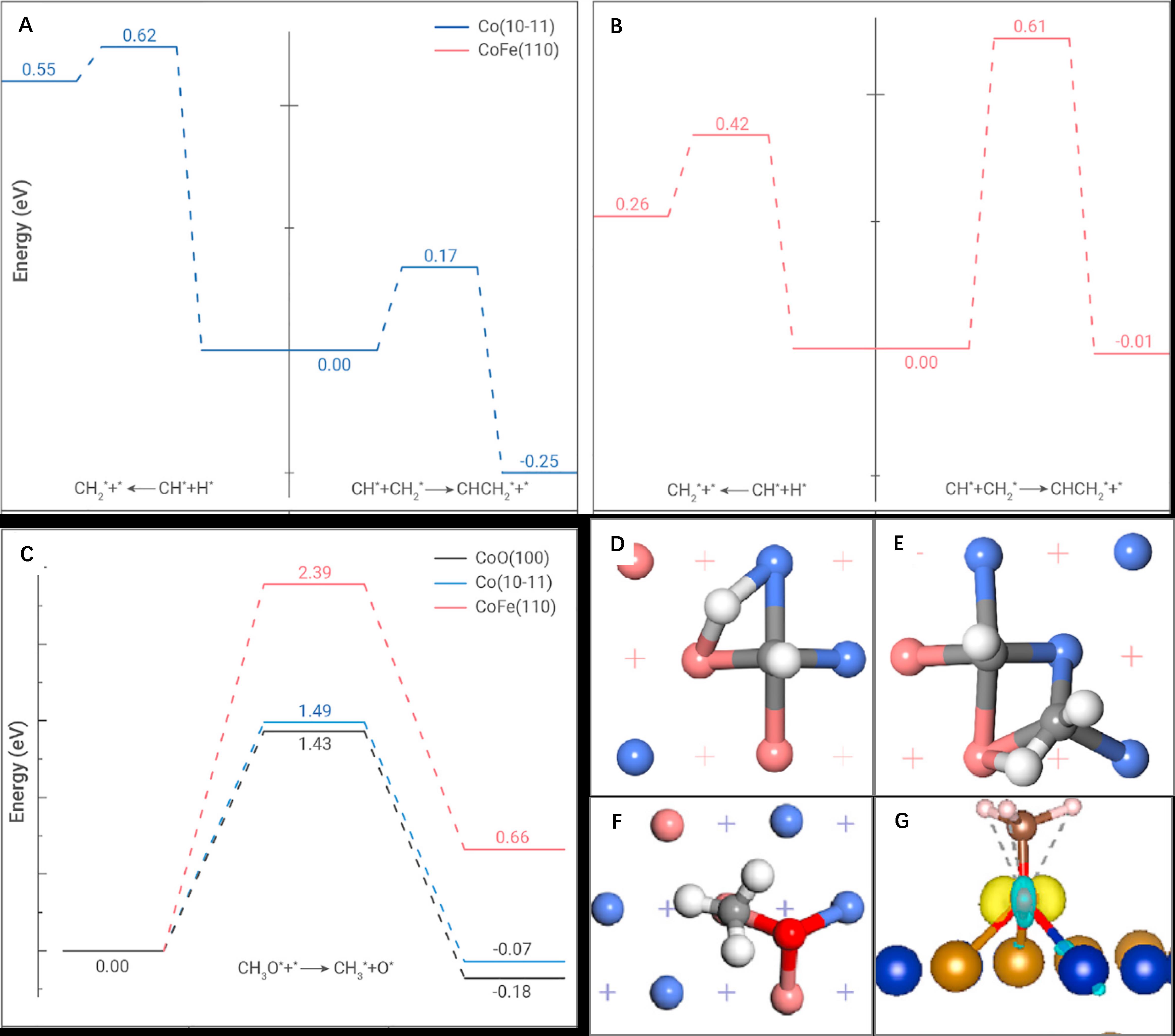
Figure 2. Density functional theory studies on CO2 hydrogenation to jet fuel. Potential energy profiles of the C-C coupling and CH* hydrogenation over (A) Co and (B) CoFe catalyst surfaces, and (C) of the CH3O* dissociation on Co, CoFe and CoO surfaces. Transition state structures of (D) C-C coupling, (E) CH* hydrogenation, and (F) CH3O* dissociation over the CoFe catalyst surface, and (G) charge density difference of the CH3O* adsorbate over the CoFe catalyst surface. (Image by SARI)
This study provides a viable technique for the highly selective synthesis of eco-friendly and carbon-neutral jet fuel from CO2, which can potentially facilitate the rational development of efficient materials for the direct hydrogenation of CO2 to advanced liquid fuels.


